
🧠 🥩 Sobre la particular historia del kuru de Papúa Nueva Guinea 🥩🧠
Un fuego de origen desconocido
Allá por 1950 llegaría a Papúa Nueva Guinea el dr. Vincent «Yis» Zigas, médico germano-estonio que residió unos años en Australia y que realizaría labores de médico rural siendo, probablemente, la única asistencia de medicina moderna en la región durante casi una década, lo que lo convirtió también en un personaje muy querido y respetado por los papúes. Zigas comenzó a escuchar rumores entre la población local de una maldición incontrolable que azotaba al pueblo Fore del Sur, afectando a mujeres y niños, y que los lugareños atribuían a la hechicería. Estos rumores lo llevaron a buscar a esta tribu de las Tierras Altas Orientales de Papúa Nueva Guinea, indagando sobre este mal, conocido por los lugareños como kuroa y que después se popularizó como kuru. Los síntomas eran, principalmente, temblores violentos, pérdida de coordinación, risa sardónica, desnutrición, estrabismo, demencia y una inevitable muerte, lo que al final terminaría acuñando el nombre pues kuru significa, literalmente, «temblar de miedo».
En 1951, el oficial australiano Arthur Carey fue el primero en utilizar ese término en un informe, después de contactar con Zigas, para describir esta nueva enfermedad. En su informe, Carey señaló que el kuru afectaba principalmente a las mujeres Fore y terminaba matándolas, extendiéndose entre el resto de los pueblos Fore del Norte, Yate y Usurufa ya en 1952 y 1953, según el matrimonio de antropólogos Ronald y Catherine Berndt. En 1953 otro oficial, John McArthur, observó también el kuru mientras patrullaba y proporcionó la primera descripción detallada de la enfermedad para enviarlo a autoridades australianas. McArthur creía que el kuru era simplemente un episodio psicosomático resultado de las prácticas de hechicería de los pueblos tribales de la región pero como ya para 1957 la enfermedad se había extendido por el tercio sureste de la isla de Nueva Guinea, los miembros de la tribu le pidieron a Charles Pfarr, un médico militar australiano en labores de servicio, que fuera al área para recabar información y poner sobre aviso, de nuevo, a las autoridades de su país por esta enfermedad que atacaba a mujeres y niños de estos entornos rurales. Hay que recordar que, a día de hoy, alrededor del 85% de la población de Papúa Nueva Guinea vive en entorno rural.
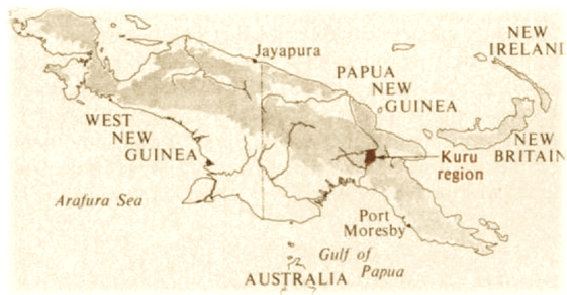
La isla de Nueva Guinea y la extensión del kuru, c. 1954.
Aunque desde el punto de vista epidemiológico se planteó que la etiología del kuru fuera infecciosa, los pacientes no presentaban signos o síntomas meningoencefalíticos (fiebre, convulsiones o coma), pleocitosis en el líquido cefalorraquídeo y, en la autopsia, no se observan manguitos perivasculares ni otros signos de la patología cerebral inflamatoria. Esta expansión del kuru, supuestamente epidémica, era inconsistente con la hipótesis genética contemporánea de Henry Bennett pero era compatible con una enfermedad infecciosa lenta. Bennet, otro médico australiano, empezó a estudiar la enfermedad a partir de los informes que le fueron llegando en los años 50.
En las autopsias se encontró con una característica muy singular: el cerebro de los afectados estaba lleno de agujeritos, como si fuera una esponja. Se conocía el fuego pero no su origen hasta que llegó Shirley Lindenbaum, antropóloga australiana, enviada por Bennet y que hizo un trabajo de campo sobre este pueblo tribal estudiando sus costumbres, rituales, creencias y entramados sociales, presentando en 1961 sus conclusiones: los Fore practicaban canibalismo ritual desde hacía más de medio siglo. Para llevar algo de esa persona dentro y para adquirir sus cualidades como herencia, el cerebro se aspiraba con una especie de sorbete a través de la nariz. Su investigación fue la clave principal para empezar a resolver el misterio dado que encontró lo que le habían encargado, esa relación entre las costumbres de los Fore y el kuru.

Lindenbaum con los Fore a su llegada en 1960.
Viscoso, pero sabroso
A finales del siglo XIX, el pueblo Fore de las Tierras Altas Orientales de Papúa-Nueva Guinea cambió sus costumbres funerarias. Los familiares fallecidos empezaron a ser cocinados y comidos, las mujeres y los niños recibían el cerebro de sus difuntos cuando se repartían las piezas. Es decir, se comían los sesos de sus seres queridos. El endocanibalismo ritualista -comer parientes como parte de un ritual de duelo en contraposición a comerse enemigos, es decir, el exocanibalismo– se practicaba no solo en el área donde empezó a darse el kuru, sino en muchos grupos circundantes de las Tierras Altas Orientales de Papúa en los que esta enfermedad nunca se desarrolló.
Cuando se considera que un cuerpo es «apto» para el consumo humano no se descarta nada excepto la vesícula biliar amarga. En la antigua huerta de caña de azúcar del difunto, los familiares maternos desmiembran el cadáver con un cuchillo de bambú y un hacha de piedra. Primero amputan las manos y los pies, luego seccionan los brazos y las piernas para desnudar los músculos. Abren el pecho y el vientre, evitando romper la vesícula biliar, cuyo amargor arruinaría la carne. Después de cortar la cabeza, fracturan el cráneo para extraer el cerebro. La médula se extrae de los huesos rotos y, a veces, los huesos pulverizados se cocinan y se comen con verduras. En los Fore del Norte, pero no del Sur, el cadáver es enterrado durante varios días, luego exhumado y comido cuando la carne ha «madurado» y los gusanos podían cocinarse «como un manjar por separado». En resumen, se comen carne, vísceras y sesos.
Los Fore recuerdan que el primer caso de kuru fue alrededor de 1920. Curiosamente, cuando apareció por primera vez el kuru, los Fore describieron sus síntomas como similares a un «casuario agitado», resaltando el gran temor que les daba acercarse al principio a las mujeres y niños Fore. Pero, al fin y al cabo, eran los suyos. Dado que los hombres no se comían el cerebro- sino otras partes del cuerpo-, no se veían afectados pero sí diezmando la población de esta tribu melanésica cada año, pues se estaban quedando las aldeas sin mujeres jóvenes ni niños. Durante los años 20 del siglo pasado se había extendido aún más, a las aldeas de los Kasokana y de los Miarasa en la región norte de los Fore, y una década más tarde había llegado al sur, en las aldeas de los Wanikanto y los Kamira. El kuru se volvió endémico en todas las aldeas en las que entró, y se volvió hiperendémico en la porción oriental de la isla. Todos los informantes nativos destacaron el origen relativamente reciente del kuru: «Esto no pasaba antes.»

Niños de los Fore con kuru. Uno de los signos de la enfermedad era la risa sardónica. C. 1970. gente Fore.
La peor persona del mundo
Sin embargo, ni los estudios ambientales ni los estudios genéticos disponibles proporcionaron pistas. Además, todos los intentos de transmitir el kuru a pequeños animales de laboratorio, o de aislar cualquier microorganismo, tuvieron éxito. Sabían por qué occurría el kuru pero no el qué lo causaba. Por qué sí a estas tribus y no a otras. En otras investigaciones de amplio alcance, como análisis genéticos exhaustivos, la búsqueda de deficiencias nutricionales o toxinas ambientales no resultó tampoco en una hipótesis defendible. Sin embargo, años antes de que Lindenbaum describriera la conexión entre el canibalismo y el kuru, Zigas le escribió una carta a otro médico australiano, John Gunther, describiéndole los signos más claros de la enfermedad:
«…Probablemente se trate de una nueva forma de encefalitis[…]comenzó con fiebre, somnolencia, dolor muscular y debilidad, dolor de cabeza […] vértigo […] caras como máscaras, flexionados brazos y muñecas, marcha inestable, trastornos oculares como diplopía, estrabismo, nistagmo, temblor de dedos y manos”
Cuentan las malas lenguas a mediados de los años 50 dicha carta cayó en manos de un oportunista llamado Carleton Gajdusek, virólogo húngaro nacionalizado estadounidense y que residió un tiempo en Australia, e insistió a Zigas para unirse a él realizando trabajo de campo entre los Fore para estudiar qué ocurría con el kuru. Allá por 1957 publicarían los primeros resultados describiendo por primera vez al kuru como una enfermedad neurodegenerativa que debutaba con ataxia progresiva e insidiosa, con dolor articular, cefalea, tos, fiebre y un tipo peculiar de balanceo al desplazarse que los locales describían como si hubiesen «perdido las rodillas». Los afectados, mayormente mujeres y niños, sufrían estrabismo y un temblor progresivo que terminaba por hacer imposible cualquier actividad, sufriendo incontinencia y postración hasta la muerte. La enfermedad era siempre mortal, generalmente en menos de un año desde la aparición de los primeros síntomas. Debemos recordar que pese a ser la primera descripción del kuru como enfermedad neurodegenerativa, no se sabía la razón de por qué ocurría hasta que Lindenbaum describió las prácticas caníbales en los años 60 y, hasta décadas después, no se sabría el agente causal de la etiología.

El kuru se cebó especialmente con los niños y mujeres Fore.
Más tarde en 1959, mientras estaba en Nueva Guinea, Gajdusek recibió una carta de un veterinario estadounidense, William Hadlow, que señalaba las analogías entre el kuru y la scrapie o tembladera ovina, una enfermedad neurodegenerativa lenta de ovejas y cabras conocida por ser endémica en el Reino Unido desde el siglo XVIII y transmitida experimentalmente a cabras desde 1936. Habiendo visto fotografías de placas de kuru en una exhibición del Wellcome Medical Museum en Londres, adjuntó una copia de una carta que señalaba esta similitud al editor de Lancet. Hadlow basó sus observaciones no solo en la presencia de placas amiloides, sino principalmente en la presencia de neuronas vacuoladas o vaciadas:
“Me ha impresionado el parecido general del kuru y un oscuro trastorno degenerativo de las ovejas llamado scrapie […] Las lesiones observadas en las cabras parecen ser notablemente parecidas a las descritas para el kuru […] Todo esto me sugiere que un enfoque experimental similar al adoptado para la tembladera podría resultar extremadamente fructífero en el caso del kuru […] porque me ha impresionado mucho la intrigante implicación, he enviado una carta a ‘The Lancet’”.
Posteriormente, a principios de la década de 1970, Gajdusek inyectó un poco de tejido cerebral que provenía de una víctima mortal de kuru en un chimpancé vivo que terminó desarrollando los síntomas, con lo cual quedó probada la transmisibilidad de esta enfermedad. Le dieron un Nobel gracias también a las investigaciones de Lindenbaum, a quién despreció siempre, y posteriormente fue condenado por abuso sexual a menores. Tela con el tío. Ya en los años 80 otro investigador estadounidense, el neurólogo e histopatólogo Stanley Prusiner, aisló el agente causal de la enfermedad, que no era un gen como creía Bennet sino una pequeña proteína bautizada como prión, que en este caso se plegaba de forma anómala. También le dieron el Nobel.
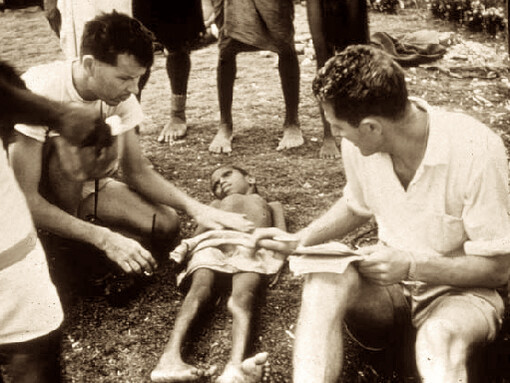
Gajdusek (izquierda) y Zigas (derecha) con un niño afectado por kuru.
A priori, es un prión
Las autoridades australianas prohibieron el canibalismo en la década de 1950, pero dado que el kuru tiene un período de incubación muy largo, de décadas incluso, a día de hoy siguen apareciendo casos en personas mayores. Esta fue la primera enfermedad priónica que demostró ser infecciosa. La enfermedad priónica humana, la enfermedad de Creutzfeldt-Jakob (ECJ), es otra encefalopatía que ocurre aproximadamente en 1 persona por millón en todas las poblaciones humanas, por lo que se planteó la hipótesis de que el kuru fue el primer caso en el que se comió a un ser humano infectado con ECJ y se propagó. Pero, ¿qué es una enfermedad priónica? Hoy sabemos que los priones, y no los virus, son responsables de las encefalopatías espongiformes transmisibles (EET), etiologías neurodegenerativas como el scrapie, la encefalopatía espongiforme bovina -también conocida como enfermedad de las vacas locas-, la enfermedad de Creutzfeldt-Jakob, la enfermedad de Gerstmann-Straussler-Scheinker. el insomnio familiar fatal en humanos y, por supuesto, el kuru.
La PRNP es el gen de la proteína priónica (PrP) y se expresa mayoritariamente en el sistema nervioso, asociándose por lo general a la memoria y a factores neuroprotectores. La enfermedad priónica es la etiología donde esta versión defectuosa de dicha proteína, el prión, se pliega mal y de alguna manera hace que las proteínas cercanas adopten la forma mal plegada, produciendo una reacción en cadena y desencadenando todos los males que conocemos en las encefalopatías espongiformes. Las proteínas son moléculas tridimensionales complejas que se pliegan en formas específicas, lo que les permite hacer su trabajo. Una proteína mal plegada puede eliminarse de la célula, pero las enfermedades priónicas provocan dicha reacción en la que otras proteínas se adaptan a esta forma mal plegada, lo que destruye progresivamente el cerebro provocando esos «huecos» en la corteza tan característicos de dichas encefalopatías. Las enfermedades priónicas son progresivas, no tienen tratamiento conocido y siempre son fatales.
En la secuencia de ADN que codifica para PRNP, el codón 129 del Cromosoma 20 en humanos, es crucial. Los homocigotos, aquellas personas con alelos idénticos de sus padres, tienen un riesgo sustancialmente mayor que los heterocigotos, aquellas que poseen dos alelos diferentes para el mismo gen. En un análisis de sangre de mujeres Fore de 50 años o más, 23 de un total de 30 eran heterocigotas en el codón 129, una cifra sustancialmente diferente de otras poblaciones. Algunos investigadores han argumentado que la epidemia de kuru ha seleccionado fuertemente la heterocigosidad que ha terminado protegiendo al pueblo Fore y esto implica que en un espacio de tiempo brevísimo hemos podido observar una adaptación en tiempo real ante una enfermedad fatal. De manera global, el patrón parece ser el mismo a lo largo del tiempo para protegernos de otras encefalopatías. La mayoría de las poblaciones tienen evidencia de una fuerte selección protectora ante el canibalismo en el pasado, lo que indica que se produjeron brotes esporádicos de enfermedades priónicas a lo largo de la prehistoria humana. Así que ahí lo tenemos, el kuru fue (es) una enfermedad devastadora para los Fore, pero los efectos evolutivos a largo plazo se mantendrán en la población durante milenios, brindando protección contra la enfermedad de Creutzfeldt-Jakob de origen natural y otras enfermedades priónicas, al igual que así ha sido a lo largo de la historia del ser humano. En distintas épocas, en distintos contextos y con distintos fuegos.
Vídeo resumen:
Referencias:
Bennett, J. H., Rhodes, F. A., & Robson, H. N. (1958). Observations on kuru. I. A possible genetic basis. Australasian annals of medicine, 7(4), 269–275.
Nelson, H. (1996). Kuru: The Pursuit of the Prize and the Cure. The Journal of Pacific History, 31(2), 178–201.
Liberski, P. P., Gajos, A., Sikorska, B., & Lindenbaum, S. (2019). Kuru, the First Human Prion Disease. Viruses, 11(3), 232.
Lindenbaum, S. (2013). Kuru Sorcery. Disease and danger in the New Guinea Highlands, 2nd edn. Paradigm Publishers, Boulder, p 224.
Lindenbaum, S. (2008). Understanding kuru: the contribution of anthropology and medicine. Philos Trans R Soc Lond Ser B Biol Sci, 363(1510):3715–3720
Prusiner, S.B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216(4542): 136-144.
Zeidler, M. (2007). Prion diseases. En: Goldman L, Ausiello D, eds. Cecil Medicine. 23ª ed. Saunders Elsevier, Filadelfia. : capítulo 442.
Zigas, V., & Gajdusek, D. C. (1957). Kuru: clinical study of a new syndrome resembling paralysis agitans in natives of the Eastern Highlands of Australian New Guinea. The Medical journal of Australia, 44(21), 745–754.
Canción recomendada / disco recomendado:
